
Align (step 2)#
Different genes evolve at different rates, and so do different regions of the gene. Therefore, we have to make the sequences comparable to one another so we can actually estimate divergence from different regions of different sequences that share a functional or structural role. And to do so, we have to align the sequences.
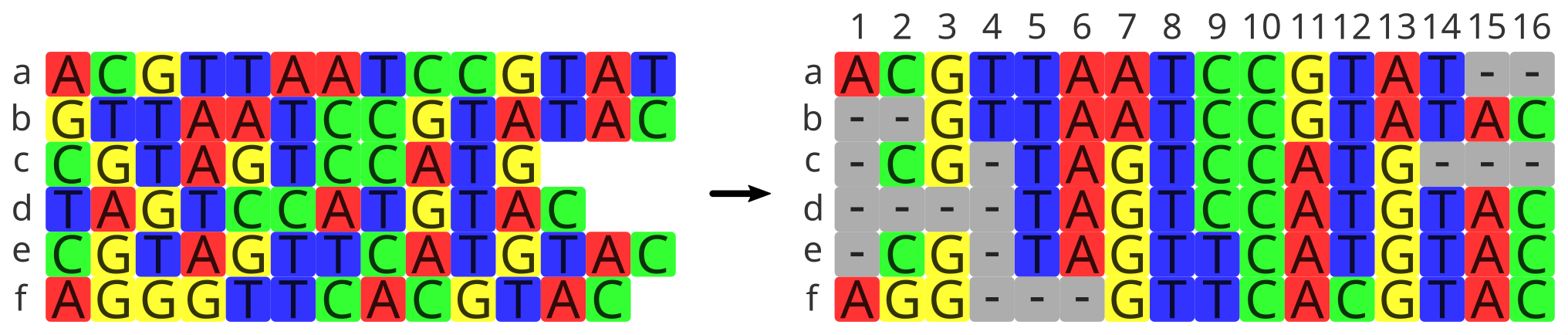
For example using MAFFT, for large dataset I normally use the default options:
mafft $FASTA > $ALIGNED
For a small dataset (<200 sequences) of different groups, I normally use:
mafft --maxiterate 1000 --localpair $FASTA > $ALIGNED
And for a small dataset (<200 sequences of similarish length) of closely related group, I normally use:
mafft --maxiterate 1000 --globalpair $FASTA > $ALIGNED
Depending on the sequences you are aligning you may want to play with the different options that MAFFT offer. I recommend playing with them and with different datasets (highly similar and highly divergent sequences) to fully understand them.
It is important to manually check the alignment in AliView (or SeaView) if you are working with recently sequenced sequences or of doubted origin. There might be some misalignment or weird stuff easy to spot due to bad quality or sequencing errors.
Other softwares offer other possibilities, for example muscle (very useful for proteins) stores ambiguities or alignment errors for downstream analysis, and clustal uses a HMM profile to generate the alignment. Further posibilities can be found at the EMBL-EBI.
It is also possible (yet requires experience) to align the sequences based on complementary regions of the hypothetical 2D structure of the given coding gene or rDNA (if known).