
Using a Maximum Likelihood (ML) approach#
Maximum Likelihood (Felsenstein, 1981) maximises model parameters (treated as constants) across different bootstraps (or “replicates”) to find a higher likelihood.
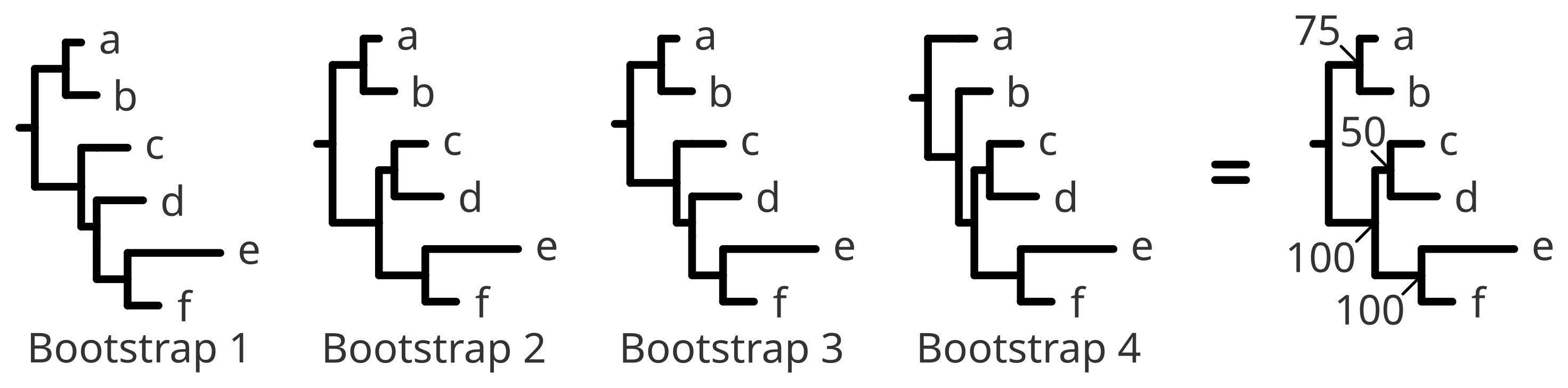
At the end, all the different replicates can be summarised into a consensus tree or simply take the best likelihood scoring tree. Either way, nodes can be annotated on the basis of how many times a given node has appeared across the different bootstraps, which is referred to as the bootstrap support (from 1 to 100).
We need now new variables:
THREADS=2 # Normally 1 thread for every 500-1000bp alignment positions works fine.
BS=100 # The number of Bootstraps
Here you have different softwares:
The first example with RAxML:
raxmlHPC-PTHREADS-SSE3 -n ${OUTPUT}_raxml-GTRgamma -s $FILE -m GTRGAMMA -p $RANDOM -x $(date +%s) -f a -N $BS -T $THREADS
or faster model and most of the times very similar output:
raxmlHPC-PTHREADS-SSE3 -n ${OUTPUT}_raxml-GTRcat -s $FILE -m GTRCAT -c 25 -p $RANDOM -x $(date +%s) -f a -N $BS -T $THREADS
With RAxML-ng you could use the Graphical User Interface option through their server: RAxML-NG, or have a look at this script for further details through the command line.
With IQ-TREE you can run modelTest, which is used to select the best substitution model fitting your data:
MEM=2GB
iqtree -s $FILE -st "DNA" -pre ${OUTPUT}_IQtree -b $BS -seed $(date +%s) -mem $MEM -nt $THREADS -wbtl
And if you know the model of evolution to be used you can add it to the command. Most of the times, the best model is the Generalised Time Reversible model (Tabaré, 1986; Lectures Math. Life Sci 17:2,57-86) with a Gamma distribution and proportion of Invariant sites for rate heterogeneity (GTR+G+I, but it also is the most complex model): -m GTR+I+G
IQ-Tree can also be used interactively in this server.
ModelTest can also be run in R, with the packages ape and phangorn (see this script for further details).
Once again, different options will address better different questions…
It is important to check the log file reporting the different analytical steps. Here you check the likelihood of the tree over the different bootstraps, the model parameters optimization or the proportion of invariant sites in the alignment, etc. Basically, you check that there is indeed an improvement and that the alignment is actually informative.